
Reaksi aldol
Reaksi aldol adalah salah satu reaksi pembentukan ikatan karbon-karbon yang penting dalam kimia organik.[1][2][3] Dalam bentuk yang umum, ia melibatkan adisi nukleofilik enolat keton ke sebuah aldehida, membentuk sebuah keton β-hidroksi, atau "aldol" (aldehida + alkohol), sebuah struktur senyawa obat-obatan yang ditemukan secara alami .[4][5][6] Kadang-kadang, produk adisi aldol melepaskan sebuah molekul air selama reaksi dan membentuk keton α,β-takjenuh. Hal ini dinamakan kondensasi aldol. Reaksi aldol ditemukan secara terpisah oleh Charles-Adolphe Wurtz[7][8][9] dan Aleksandr Porfyrevich Borodin pada tahun 1872. Borodin mengamati dimerisasi aldol 3-hidroksibutanal dari asetaldehida di bawah kondisi asam. Reaksi aldol digunakan secara meluas pada produksi komoditi kimia berskala besar seperti pentaeritritol[10] dan pada industri farmasi untuk sintesis obat-obatan yang beroptik murni. Sebagai contoh, lintasan awal Pfizer untuk obat sakit jantung Lipitor (INN: atorvastatin) yang terdaftar pada tahun 1996 menggunakan dua reaksi aldol, mengizinkan produksi obat berkuantitas skala multigram.[11][12]

Pola struktur aldol sangat umum terdapat pada poliketida, sebuah kelas produk alami yang darinya banyak obat-obatan diturunkan, meliputi immunosupresan manjur FK506, antibiotik tetrasiklina, dan agen antijamur amfoterisin B. Riset yang ekstensif terhadap reaksi aldol telah menghasilkan metode-metode reaksi yang sangat efisien, yang memperbolehkan sinstesis banyak poliketida. Tanpa metode ini, sintesis poliketida akan sangat sulit.[13] Hal ini sangatlah penting karena banyak poliketida, bersama dengan molekul-molekul aktif biologis lainnya, ditemukan secara alami dalam jumlah yang sangat sedikit untuk diinvestigasi lebih lanjut. Sintesis dari senyawa-senyawa tersebut yang pernah dianggap tidak mungkin dapat dilakukan sekarang secara rutin dalam skala laboratorium dan mendekati viabilitas ekonomi pada skala yang lebih besar pada kasus-kasus tertentu, misalnya pada agen anti-tumor yang sangat aktif, diskodermolida. Di bidang biokimia, reaksi aldol adalah salah satu langkah kunci dalam glikolisis, dimana reaksi ini dikatalisasi oleh enzim aldolase.
Reaksi aldol sangat penting dalam sintesis organik karena ia menghasilkan produk dengan dua pusat stereogenik yang baru (pada karbon -α dan -β aduk aldol, ditandai dengan tanda bintang pada gambar di atas). Metode modern sekarang ini mengizinkan kontrol pada konfigurasi relatif dan absolut pusat-pusat ini. Hal ini sangatlah penting dalam sintesis obat-obatan karena molekul-molekul dengan konektivitas struktur yang sama namun stereokimia yang berbeda sering kali memiliki sifat-sifat kimia dan biologi yang jauh berbeda.
Berbagai macam nukleofil dapat digunakan dalam reaksi aldol, meliputi enol, enolat, dan enol eter dari keton, aldehida, dan senyawa-senyawa karbonil lainnya. Pasangan elektrofiliknya biasanya adalah sebuah aldehida, walaupun terdapat juga variasi lainnya, seperti pada reaksi Mannich. Ketika nukleofil dan elektrofilnya berbeda (biasanya begitu), reaksi ini dikenal sebagai reaksi aldol silang (berlawanan dengan pembentukan dimer pada dimerisasi aldol).

Kedua labu tersebut direndam dalam penangas es kering/aseton (-78 °C). Temperatur diawasi oleh termokopel (kawat sebelah kiri)
Mekanisme-mekanisme reaksi
Reaksi aldol dapat berjalan melalui dua mekanisme yang secara mendasar berbeda. Senyawa karbonil, seperti aldehida dan keton, dapat diubah menjadi enol ataupun enol eter. Senyawa-senyawa yang bersifat nukleofil pada karbon-α ini dapat menyerang karbonil terprotonasi yang sangat reaktif. Ini merupakan "mekanisme enol". Sebagai asam karbon, senyawa-senyawa karbonil juga dapat terdeprotonasi membentuk enolat yang lebih nukleofil daripada enol maupun enol eter dan dapat secara langsung menyerang elektrofil. Biasanya elektrofil tersebut adalah aldehida karena keton pada umumnya kurang reaktif dibandingkan aldehida. Ini merupakan "mekanisme enolat".
Jika kondisi reaksi sangat "kuat" (misalnya terdapat NaOMe, MeOH, refluks), kondensasi dapat terjadi. Hal ini dapat dihindari jika menggunakan reagen-reagen yang lemah dan temperatur yang rendah (misalnya dengan kondisi dalam LDA (basa kuat), THF, -78 °C). Walaupun adisi aldol bisanya akan berjalan sampai penuh, reaksi ini tidaklah takreversibel, karena jika aduk (adduct) aldol diberikan basa kuat biasanya akan mengakibatkan pembelahan (cleavage) retro-aldol (menghasilkan senyawa semula). Kondensasi aldol bersifat takreversibel.
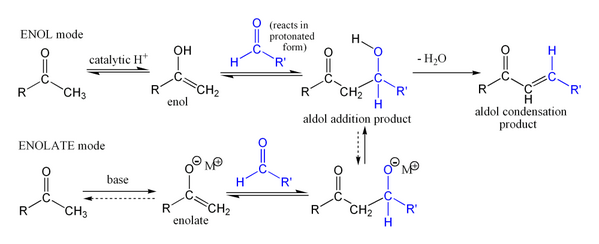
Gambaran umum dari reaksi Aldol

Mekanisme enol
Ketika katalis asam digunakan, langkah awal dari mekanisme reaksi melibatkan tautomerisasi yang dikatalisasi oleh asam pada senyawa karbonil membentuk enol. Asam juga memiliki peran mengaktivasi gugus karbonil molekul lain dengan melakukan protonasi, menjadikan molekul tersebut bersifat elektrofilik. Enol bersifat nukleofilik pada karbon-α, sehingga mengakibatkan ia dapat menyerang senyawa karbonil yang terprotonasi, menghasilkan aldol setelah deprotonasi. Biasanya akan terjadi dehidrasi dan menghasilkan senyawa karbonil takjenuh. Gambar di bawah ini menunjukkan swakondensasi aldehida yang dikatalisasikan oleh asam:
- Mekanisme aldol dengan katalis asam

Mechanism for acid-catalyzed aldol reaction of an aldehyde with itself

- Dehidrasi dengan katalis asam
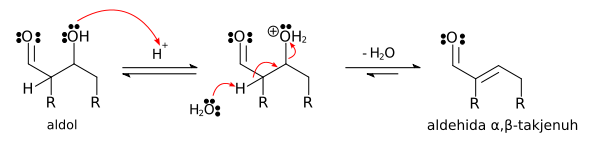
Mechanism for acid-catalyzed dehydration of an aldol

Mekanisme enolat
Jika katalis yang digunakan merupakan basa yang moderat seperti ion hidroksida atau sebuah alkoksida, reaksi aldol akan terjadi melalui serangan nukleofilik oleh enolat pada gugus karbonil molekul lain yang terstabilisasi oleh resonansi. Produk reaksi ini adalah garam alkoksida dari produk aldol. Aldol itu sendiri akan terbentuk dan dapat mengalami dehidrasi, menghasilkan senyawa karbonil takjenuh. Gambar di bawah ini menunjukkan mekanisme sederhana untuk reaksi swakondensasi aldehida yang dikatalisasikan oleh basa.
Reaksi aldol dengan katalis basa (diperlihatkan menggunakan −OCH3 sebagai basa)

Simple mechanism for base-catalyzed aldol reaction of an aldehyde with itself

Dehidrasi dengan katalis basa (kadang-kadang ditulis sebagai satu langkah tunggal)

Simple mechanism for the dehydration of an aldol product

Walaupun basa yang diperlukan hanyalah sedikit (sebagai katalis), namun biasanya digunakan basa kuat seperti LDA atau NaHMDS dengan kadar yang setara secara stoikimetri. Dengan demikian, pembentukan enolat menjadi takreversibel, dan produk aldol tidak akan terbentuk sampai alkoksi logam dari produk aldol terprotonasi pada langkah reaksi terpisah.
Model Zimmerman-Traxler
Pada tahun 1957, Zimmerman dan Traxler mengajukan bahwa beberapa reaksi aldol memiliki "keadaan transisi beranggota enam yang memiliki konformasi kursi."[14] Ini dikenal sebagai model Zimmerman-Traxler. E-enolat menghasilkan produk anti, sedangkan Z-enolat menghasilkan produk sin. Faktor-faktor yang mengontrol selektivitas ini adalah adanya preferensi meletakkan substituen secara ekuatorial pada keadaaan transisi beranggota enam dan penghindaran interaksi sin-pentana.[15] E dan Z merujuk pada hubungan stereokimia cis-trans antara enolat oksigen yang membawa ion lawan positif dengan gugus berprioritas paling tinggi pada karbon alfa. Sebenarnya, hanya beberapa logam seperti litium dan boron saja yang mengikuti model Zimmerman-Traxler ini. Sehingga pada beberapa kasus, stereokimia hasil produk reaksi tidak bisa diprediksi.
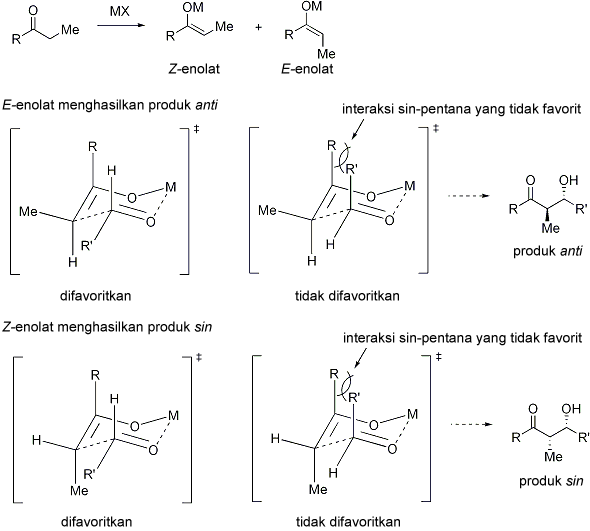
The Zimmerman-Traxler Model

Kontrol pada reaksi aldol
Masalah
Misalkan terdapat suatu reaksi hipotetis:

Dalam reaksi ini, dua keton yang tidak simetris akan dikondensasikan menggunakan natrium etoksida. Kebasaan dari natrium etoksida tidak bisa mengakibatkan deprotonasi penuh pada kedua keton tersebut, namun dapat menghasilkan sejumlah kecil natrium enolat dari kedua keton. Hal ini berarti bahwa selain berpotensi sebagai elektrofil aldol, kedua keton ini juga dapat berperan sebagai nukleofil melalui natrium enolat masing-masing. Dua elektrofil dan dua nukleofil memiliki kemungkinan empat hasil produk:

Sehingga, jika seseorang hanya ingin mendapatkan salah satu produk reaksi silang ini, maka harus dilakukan "kontrol" pada adisi aldol ini.
Keasaman
Jika satu pasangan ternyata lebih asam dari lainnya, maka secara otomatis terjadi kontrol reaksi. Proton yang paling asam akan ditarik oleh basa dan enolat akan terbentuk. Jenis kontrol ini hanya berjalan jika perbedaan keasaman cukup besar dan tidak terdapat basa yang berlebih. Jika hanya satu reaktan yang memiliki proton asam, maka hanya molekul ini sajalah yang akan membentuk enolat. Sebagai contoh, adisi dari dietil malonat dengan benzaldehida hanya menghasilkan satu produk:

Dalam kasus ini, proton metilena dari malonat akan dideprotonasi lebih banyak secara kuantitatif oleh natrium etoksida, membentuk natrium enolat. Oleh karena benzaldehida tidak mempunyai proton alfa yang asam, hanya terdapat satu kemungkinan kombinasi nukleofil-elektrofil, sehingga kontrol reaksi bisa dicapai. Perlu diperhatikan pula pendekatan ini menggabungkan dua elemen kontrol: peningkatan keasaman proton alfa pada nukleofil dan kurangnya proton alfa pada elektrofil.
Urutan penambahan
Salah satu solusi dari masalah kontrol reaksi ini adalah dengan membentuk enolat dari salah satu pasangan terlebih dahulu, kemudian menambahkan pasangan lainnya di bawah kontrol kinetik.[16] Kontrol kinetik berarti bahwa reaksi adisi aldol haruslah secara signifikan lebih cepat daripada reaksi retro-aldol (kebalikan reaksi). Agar pendekatan ini berhasil, dua kondisi reaksi lainnya juga haruslah dipenuhi, yaitu haruslah mungkin untuk sebuah pasangan menghasilkan enolat secara kuantitatif dan reaksi aldol yang berjalan secara signifikan harus lebih cepat daripada transfer enolat dari satu pasangan ke pasangan lainnya. Kondisi kontrol kinetik yang umumnya dilakukan melibatkan pembentukan enolat dari sebuah keton dengan LDA pada -78 °C, diikuti dengan penambahan aldehida yang lambat.
Enolat
Pembentukan
Enolat dapat dibentuk dengan menggunakan basa kuat ("kondisi keras") ataupun menggunakan asam Lewis dengan basa lemah ("kondisi lunak"):

Agar deprotonasi dapat terjadi, ikatan sigma alfa-C-H harus dapat bertumpang tindih dengan orbital pi* karbonil:

Geometri
Pembentukan enolat di bawah berbagai macam kondisi telah dikaji secara ekstensif. Adalah mungkin, pada kebanyakan kasus, untuk menghasilkan geometri enolat yang kita inginkan:[17]

Untuk keton, kebanyakan kondisi enolisasi menghasilkan Z enolat, sedangkan untuk ester, kebanyakan kondisi enolisasi menghasilkan E enolat. Penambahan HMPA diketahui membalikkan stereoselektivitas deprotonasi.

Pembentukan enolat yang stereoselektif dapat dirasionalkan dengan menggunakan model Ireland,[18][19][20][21] walaupun validitasnya masih dipertanyakan. Pada kebanyakan kasus, tidaklah diketahui apakah zat antaranya bersifat monomerik atau oligomerik; walaupun demikian, model Ireland masih merupakan model yang berguna untuk memahami enolat.

Dalam model Ireland, deprotonasi diasumsikan berjalan melalui keadaan transisi beranggota enam. Dua substituen yang lebih besar pada elektrofil (dalam kasus di atas, metil lebih besar daripada proton) mengadopsi disposisi ekuatorial, menghasilkan preferensi E enolat. Model tersebut gagal pada kebanyakan kasus; sebagai contoh, jika campuran pelarut diganti dari THF menjadi 23% HMPA-THF, geometri enolat berbalik tanpa bisa dijelaskan.
Enolat kinetik vs. termodinamik
Jika keton taksimetris diberikan basa, ia mempunyai potensi membentuk dua enolat yang bersifat regioisomer (dengan menghiraukan geometri enolat). Sebagai contoh:

Enolat yang ter-trisubstitusi dianggap sebagai enolat kinetik, sedangkan enolat yang ter-tetrasubstitusi dianggap sebagai enolat termodinamik. Hidrogen alfa yang terdeprotonasi membentuk enolat kinetik kurang terhalang, sehingga terdeprotonasi lebih cepat. Secara umum, olefin yang ter-tetrasubstitusi lebih stabil daripada olefin yang ter-trisubtitusi oleh karena stabilisasi hiperkonjugasi. Rasio dari regioisomer enolat ini sangat dipengaruhi oleh pilihan basa yang digunakan. Untuk contoh di atas, kontrol kinetik dapat dilakukan dengan menggunakan LDA pada -78 °C, menghasilkan selektivitas 99:1 untuk enolat kinetik:termodinamik, sedangkan kontrol termodinamik dapat dilakukan dengan menggunakan trifenilmetillitium pada suhu kamar, menghasilkan selektivitas 10:90.
Secara umum, enolat kinetik lebih difavoritkan pada kondisi temperatur yang rendah, ikatan logam-oksigen yang relatif ion, dan deprotonasi cepat menggunakan basa yang kuat dan terhalang, yang sedikit berlebihan, sedangkan enolat termodinamik lebih difavoritkan pada kondisi temperatur yang lebih tinggi, ikatan logam-oksigen yang relatif kovalen, dan waktu kesetimbangan yang lebih lama untuk deprotonasi dengan menggunakan basa kuat yang kadarnya sedikit berlebih dari jumlah sub-stoikiometri reaksi. Penggunaan jumlah sub-stoikiometri basa mengizinkan sebagian kecil senyawa karbonil yang tidak terenolisasi menyeimbangkan enolat menjadi regioisomer termodinamik dengan berperan sebagai ulang alik proton (proton shuffle).
Stereoselektivitas
Reaksi aldol sangatlah berguna karena dua pusat stereogenik yang baru dapat dihasilkan dalam satu reaksi. Riset secara ekstensif telah dilakukan untuk memahami mekanisme dan meningkatkan selektivitas reaksi ini. Konvensi sin/anti biasanya digunakan untuk menandai stereokimia relatif pada karbon-α dan -β.

Konvensi ini berlaku ketika nukleofil propionat (atau yang berderajat lebih tinggi) diadisi ke aldehida. Gugus R keton dan gugus R' aldehida diposisikan menjadi pola "zig zag" pada bidang kertas, dan disposisi dari stereopusat yang terbentuk disebut sin atau anti tergantung apakah kedua stereopusat yang terbentuk tersebut berada pada sisi yang sama atau berlawanan.
Naskah lama menggunakan tatanama eritro-treo yang dikenal dari kimia karbohidrat.
E vs. Z enolat
Tidak terdapat perbedaan yang besar pada tingkat stereoinduksi (stereoinduction) yang terpantau pada E enolat dengan Z enolat:[17]


Ion logam
Kation logam enolat memainkan peran yang besar dalam penentuan tingkat stereoselektivitas suatu reaksi aldol. Boron sering digunakan karena panjang ikatnya secara signifikan lebih pendek daripada logam-logam lainnya seperti litium, aluminium, ataupun magnesium. Sebagai contoh, ikatan boron-karbon memiliki panjang 1,4-1,5 Å dan ikatan boron-oksigan 1.5–1.6 Å. Sedangkan ikatan logam-karbon dan logam-oksigen secara umum memiliki panjang ikat 1,9-2,2 Å dan 2,0–2,2 Å secara berurutan. Hal ini memberikan efek "memperketat" keadaan transisi reaksi:[22]

Stereoselektivitas: stereopusat alfa pada enolat
Reaksi aldol dapat menunjukkan "stereokontrol yang berdasarkan substrat", yaitu kiralitas pada kedua reaktannya dapat memengaruhi stereokimia hasil reaksi. Jika enolat mempunyai sebuah stereopusat (stereocenter) pada posisi alfa, streokontrol yang baik dapat dicapai.

Pada kasus sebuah E enolat, unsur kontrol yang dominan adalah 1,3-terikan alilik (allylic 1,3-strain), sedangkan pada kasus sebuah Z enolat, unsur kontrol yang dominan adalah penghindaran interaksi 1,3-diaksial. Model umum ditunjukkan pada gambar di bawah:

Agar lebih jelas, stereopusat pada enolat telah diepimerisasikan; dalam kenyataannya, diastereomuka (diastereoface) yang berseberangan dari aldehida dapat diserang. Pada kedua kasus, stereomer 1,3-sin difavoritkan. Terdapat banyak contoh stereokontrol sejenis ini:[23]

Stereoselektivitas: stereopusat alfa pada elektrofil
Ketika enolat menyerang aldehida dengan sebuah stereopusat alfa, stereokontrol yang baik juga dimungkinkan. Pemantauan yang umum pada E enolat menunjukkan seleksi diastereomuka Felkin, sedangkan Z enolat menunjukan selektivitas anti-Felkin. Model umum selektivitas ini [24][25] ditunjukkan pada gambar di bawah:

Oleh karena Z enolat harus bereaksi melalui sebuah keadaan transisi yang kedua-duanya memiliki interaksi sin-pentana ataupun rotamer anti-Felkin yang bersifat mengurangi stabilitas senyawa, Z enolat memiliki diastereoselektivitas yang lebih rendah dalam kasus ini. Beberapa contoh diberikan pada gambar di bawah:[26][27]

Stereoselektivitas: model gabungan untuk stereoinduksi
Jika kedua enolat dan aldehida memiliki kiralitas pada awalnya, maka hasil reaksi aldol "stereodiferensiasi ganda" (double stereodifferentiating) dapat diprediksi menggunakan model stereokimia gabungan yang melibatkan bias muka enolat (enolate facial bias), geometri enolat, dan bias muka aldehida.[28] Beberapa contoh dari aplikasi model diberikan pada gambar di bawah ini:[27]

Kimia oksazolidinon Evans
Sintesis organik modern sekarang ini memerlukan sintesis senyawa dalam bentuk enantiomurni. Oleh karena reaksi adisi aldol menghasilkan dua stereopusat yang baru, terdapat paling banyak empat stereoisomer yang dihasilkan.

Terdapat banyak metode yang telah dikembangkan untuk mengontrol stereokimia relatif (sin ataupun anti) ataupun stereokimia absolut reaksi aldol.

Metode yang paling umum dipakai adalah metode oksazolidinon asil Evans.[29][30] Dikembangkan pada akhir tahun 1970-an dan awal 1980-an oleh David A. Evans dkk., metode ini bekerja dengan cara membuat enolat kiral untuk sementara waktu dengan melekatkan sebuah zat bantu kiral (chiral auxiliary). Kiralitas dari zat bantu ini kemudian ditransfer ke aduk aldol dengan reaksi aldol diastereoselektif. Setelah pembantu tersebut dilepaskan, stereoisomer yang kita inginkan akan muncul.

Dalam kasus metode Evans ini, zat bantu kiral yang dilekatkan adalah oksazolidinon, dan senyawa karbonil yang dihasilkan merupakan sebuah imida. Berbagai macam oksazolidinon tersedia dalam kedua bentuk enantiomer. Oksazolidon ini memakan biaya sekitar $10–$20 dollar AS per gram.

Asilasi oksazolidinon merupakan prosedur yang mudah dilakukan, dan secara informal dirujuk sebagai "loading done". Z-enolat, yang menghasilkan aduk sin-aldol, dapat dibentuk dengan mudah menggunakan enolisasi lunak yang diperantarai boron:[31]

Sering kali, diastereomer dapat diperoleh dengan satu kali kristalisasi aduk aldol. Sayangnya, aduk anti-aldol tidak bisa didapatkan dengan mudah menggunakan metode Evans. Walaupun metode ini mahal dan memiliki batasan yang hanya memberikan aduk sin, metode ini sering dipilih oleh karena dapat diandalkan, mudah digunakan, dan serbaguna. Terdapat banyak metode pembelahan pembantu kiral ini:[32]

Setelah pembentukan imida dilakukan, baik reaksi adisi aldol yang sin- maupun anti-selektif dapat dilakukan, mengijinkan "perakitan" tiga dari empat stereolarik (Bahasa Inggris: stereoarray): sin-selektif:[33] dan anti-selektif:[34]

Pada reaksi sin-selektif, kedua metode enolisasi memberikan Z enolat, seperti yang diharapkan; namun stereokimia hasil akhir reaksi ini dikontrol oleh stereokimia metil daripada kiralitas oksazolidinon. Metode-metode yang dijelaskan di atas mengijinkan perakitan stereokselektif poliketida, sebuah kelas produk alami yang sering mempunyai retron aldol.
Kimia aldol modern
Metodologi mutakhir sekarang ini mengijinkan dilakukannya berbagai variasi reaksi aldol, sering kali dengan menggunakan sejumlah kecil ligan kiral. Ketika pada reaksi digunakan sejumlah kecil ligan yang secara enantiomer murni untuk menginduksi pembentukan produk yang juga secara enantiomer murni, reaksi ini umumnya diistilahkan sebagai "katalitik, asimetrik".
Reaksi aldol asetat
Keterbatasan dari pendekatan zat bantu kiral yang disebutkan sebelumnya ada pada kegagalan imida N-asetil untuk bereaksi secara selektif. Pendekatan awal menggunakan gugus tioeter sementara.[32][35]

Reaksi aldol Mukaiyama
Reaksi aldol Mukaiyama adalah adisi nukleofilik silil enol eter ke aldehida yang dikatalisasi oleh asam Lewis seperti boron triflourida atau titanium klorida.[36][37] Reaksi aldol Mukaiyama tidak mengikuti model Zimmerman-Traxler.
Metode ini bekerja untuk aldehida alifatik yang tidak bercabang, yang sering kali merupakan elektrofil buruk untuk proses katalitik asimetrik. Kemungkinan ini disebabkan oleh pembedaan (differentiation) elektronik dan sterik di antara enantiomuka masing-masing.

Proses aldol Mukaiyama vinilog analog juga dapat diubah menjadi katalitik dan asimetrik. Contoh di bawah ini bekerja dengan efisien untuk aldehida aromatik (namun tidak pada alifatik) dan mekanismenya diduga melibatkan dienolat kiral yang terikat pada logam.[38][39]

Aldol tiazolidinetion Crimmins
Terdapat pula versi zat bantu Evans yang lainnya, yaitu tiazolidinetion Crimmins.[40][41]
Hasil reaksi, diastereoselektivitas, dan enantioselektivitas reaksi ini pada umumnya tinggi, walaupun tidak setinggi metode Evans. Tidak seperti zat bantu Evans, tiazolidinetion dapat menjalankan reaksi aldol asetat (ref: Crimmins, Org. Lett. 2007, 9(1), 149–152.) dan dapat menghasilkan aduk "sin Evans" maupun "sin non-Evans" hanya dengan mengubah kadar (-)-sparteina. Reaksi ini dipercaya berjalan via keadaan transisi beranggota enam yang terikat dengan titanium, memiliki analogi yang sama dengan keadaan transisi zat bantu Evans.

Reaksi aldol organokatalitik
Terdapat perkembangan baru dengan menggunakan katalis amina sekunder kiral. Amina sekunder ini akan membentuk enamina fana (transient) ketika terekspos dengan keton, yang dapat bereaksi secara enantioselektif dengan elektrofil aldehida yang tepat. Ini dikenal sebagai katalisis enamina, sejenis organokatalisis, karena katalis ini keseluruhannya berasal dari molekul organik yang kecil. Pada contoh di bawah ini, prolina secara efisien mengkatalisasi siklisasi triketon:

Reaksi ini dikenal sebagai reaksi Hajos-Parrish[42][43] (juga dikenal sebagai reaksi Hajos-Parrish-Eder-Sauer-Wiechert)[44]. Reaksi Hajos-Parrish ini hanya membutuhkan sejumlah kecil prolina (3 mol%). Tidak terdapat reaksi latar akiral karena zat antara enamina fana lebih nukleofilik daripada enol keton induk. Strategi ini sangatlah berguna karena ia menawarkan cara yang sederhana untuk mendapatkan enantioselektivitas reaksi tanpa menggunakan logam transisi yang beracun atau mahal.
Menariknya, reaksi aldol yang dikatalisasi oleh prolina tidak menunjukkan efek-efek non-linear (enantioselektivitas produk secara langsung proposional dengan kemurnian enantiomer katalis). Dengan menggabungkan bukti penandaan isotopik dan kajian komputasi, mekanisme reaksi yang diajukan untuk reaksi aldol yang dikatalisasi oleh prolina adalah sebagai berikut:[45]

Strategi ini mengizinkan reaksi aldol silang antara dua aldehida. Secara umum, reaksi silang aldol aldehida menantang karena ia dapat berpolimerisasi dengan mudah atau bereaksi secara tidak selektif, memberikan campuran statistis produk (statistical mixture). Contoh:[46]

Berlawanan dengan preferensi aduk sin yang umumnya terpantau pada adisi aldol berbasis enolat, adisi aldol yang di-organokatalisasi ini anti selektif. Pada kebanyakan kasus, kondisi organokatalitik ini cukup lunak untuk menghindari polimerisasi. Namun, selektivitas memerlukan pompa semprit (syringe) yang lambat untuk mengontrol penambahan pasangan elektrofilik yang diinginkan karena kedua pasangan reaksi ini umumnya memiliki proton yang bisa dienolisasi. Jika satu aldehida tidak memiliki proton yang bisa dienolisasi atau pencabangan alfa atau beta, kontrol tambahan dapat dicapai.
Demonstrasi elegan dari reaksi aldol organokatalitik asimetrik yang hebat ini diperlihatkan oleh MacMillan dkk. pada tahun 2004 pada sintesis karbohidrat yang diproteksi secara diferensial. Ketika metode sintesis tradisional menyelesaikan sintesis heksosa menggunakan berbagai variasi strategi proteksi-deproteksi iteratif yang memerlukan 8-14 langkah, organokatalis dapat mengakses banyak substrat yang sama menggunakan protokol dua langkah yang efisien, melibatkan dimerisasi yang dikatalisasi oleh prolina pada alfa-oksialdehida, diikuti dengan siklisasi aldol Mukaiyama tandem.

Dimerisasi aldol alfa-oksialdehida mengharuskan aduk aldol, yang sendiri merupakan aldehida, bersifat inert untuk reaksi aldol lebih lanjut.[47] Kajian awal menunjukkan bahwa aldehida yang membawa substituen alfa-alkiloksi atau alfa-sililoksi cocok untuk reaksi ini, sedangkan aldehida yang membawa gugus penarik elektron seperti asetoksi tidak reaktif. Produk eritrosa yang terlindungi dapat kemudian diubah menjadi empat jenis gula yang dimungkinkan dengan adisi aldol Mukaiyama, diikuti dengan pembentukan laktol. Ini memerlukan diastereokontrol yang tepat pada adisi aldol Mukaiyama dan mengharuskan produk ion sililoksikarbenium memiliki preferensi untuk bersiklisasi daripada mengalami reaksi aldol lebih lanjut. Pada akhirnya, glukosa, manosa, dan alosa berhasil disintesis:

Adisi aldol "langsung"
Pada adisi aldol yang biasanya, senyawa karbonil diprotonasi dan membentuk enolat. Enolat kemudian ditambahkan ke sebuah aldehida atau keton, membentuk alkoksida yang kemudian terprotonasi pada langkah selanjutnya. Metode yang lebih superior akan menghindari urutan deprotonasi-aldol-protonasi ini dan lebih memilih "adisi aldol langsung". Hal ini dikarenakan pada proses tersebut, adisi aldol menghasilkan alkoksida yang lebih basa daripada senyawa awal, menghindari penggantian katalis:

Satu pendekatan yang baru-baru ini didemonstrasikan oleh Evans adalah dengan mensililasi aduk aldol:[48]

Metode ini lebih hemat dan secara industri lebih berguna daripada prosedur umumnya yang berbasis enolat. Baru-baru ini, pendekatan biomimetik oleh Shair menggunakan beta-tioketoasam sebagai nukleofil.[49] Gugus ketoasamnya di-dekarboksilasi secara in situ (ligan kiralnya adalah bisoksazolina). Menariknya, aldehida aromatik dan alifatik bercabang merupakan substrat yang buruk.

Lihat pula
- Reaksi Aldol-Tishchenko
- Reaksi Baylis-Hillman
- Reaksi Ivanov
- Reaksi Reformatsky
- Reaksi Cannizzaro
- Asam aldonat
- Kondensasi Knoevenagel
Referensi
- ^ Wade, L. G. (2005). Organic Chemistry. Upper Saddle River, New Jersey: Prentice Hall. hlm. 1056–1066. ISBN 0-13-236731-9.
- ^ Smith, M. B.; March, J. (2001). Advanced Organic Chemistry. New York: Wiley Interscience. hlm. 1218–1223. ISBN 0-471-58589-0.
- ^ Mahrwald, R. (2004). Modern Aldol Reactions, Volumes 1 and 2. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA. hlm. 1218–1223. ISBN 3-527-30714-1.
- ^ Heathcock, C. H. (1991). Comp. Org. Syn. Oxford: Pergamon. hlm. 133–179. ISBN 0-08-040593-2.
- ^ Mukaiyama T. (1982). "The Directed Aldol Reaction". Org. React. 28: 203–331.
- ^ Paterson, I. (1988). "New Asymmetric Aldol Methodology Using Boron Enolates". Chem. Ind. 12: 390–394.
- ^ Wurtz, C. A. (1872). Bull. Soc. Chim. Fr. 17: 436–442.
- ^ Wurtz, C. A. (1872). "Ueber einen Aldehyd-Alkohol". J. Prakt. Chemie. 5 (1): 457–464. doi:10.1002/prac.18720050148.
- ^ Wurtz, C. A. (1872). "Sur un aldéhyde-alcool". Comp. Rend. 74: 1361.
- ^ Mestres R. (2004). "A green look at the aldol reaction". Green Chemistry. 12: 583–603. doi:10.1039/b409143b.
- ^ M. Braun, R. Devant (1984). "(R) and (S)-2-acetoxy-1,1,2-triphenylethanol - effective synthetic equivalents of a chiral acetate enolate". Tetrahedron Letters. 25: 5031–4. doi:10.1016/S0040-4039(01)91110-4.
- ^ Jie Jack Li; et al. (2004). Contemporary Drug Synthesis. Wiley-Interscience. hlm. 118–. ISBN 0-471-21480-9.
- ^ Schetter, B., Mahrwald, R. (2006). "Modern Aldol Methods for the Total Synthesis of Polyketides". Angew. Chem. Int. Ed. 45: 7506–7525. doi:10.1002/anie.200602780.
- ^ Zimmerman, H. E.; Traxler, M. D. (1957). "The Stereochemistry of the Ivanov and Reformatsky Reactions. I". J. Am. Chem. Soc. 79: 1920–1923. doi:10.1021/ja01565a041.
- ^ Heathcock C. H., Buse, C. T., Kleschnick W. A., Pirrung M. C., Sohn J. E., Lampe, J. (1980). "Acyclic stereoselection. 7. Stereoselective synthesis of 2-alkyl-3-hydroxy carbonyl compounds by aldol condensation". J. Org. Chem. 45: 1066–1081. doi:10.1021/jo01294a030.
- ^ Bal, B.; Buse, C. T.; Smith, K.; Heathcock, C. H. Org. Syn., Coll. Vol. 7, p.185 (1990); Vol. 63, p.89 (1985). (Article)
- ^ a b Brown H. C., Dhar R. K., Bakshi R. K., Pandiarajan P. K., Singaram B. (1989). "Major effect of the leaving group in dialkylboron chlorides and triflates in controlling the stereospecific conversion of ketones into either E- or Z-enol borinates". J. Am. Chem. Soc. 111: 3441–3442. doi:10.1021/ja00191a058.
- ^ Ireland, R. E.; Willard, A. K. (1975). "The stereoselective generation of ester enolates". Tetrahedron Lett. 16 (46): 3975–3978. doi:10.1016/S0040-4039(00)91213-9.
- ^ Narula, A. S. (1981). "An analysis of the diastereomeric transition state interactions for the kinetic deprotonation of acyclic carbonyl derivatives with lithium diisopropylamide". Tetrahedron Lett. 22 (41): 4119–4122. doi:10.1016/S0040-4039(01)82081-5.
- ^ Ireland, R. E.; Wipf, P.; Armstrong, J. D. (1991). "Stereochemical control in the ester enolate Claisen rearrangement. 1. Stereoselectivity in silyl ketene acetal formation". J. Org. Chem. 56: 650–657. doi:10.1021/jo00002a030.
- ^ Xie L., Isenberger K. M., Held G., Dahl, L. M. (1997). "Highly Stereoselective Kinetic Enolate Formation: Steric vs Electronic Effects". J. Org. Chem. 62: 7516–7519. doi:10.1021/jo971260a.
- ^ Evans D. A., Nelson J. V., Vogel E., Taber T. R. (1981). "Stereoselective aldol condensations via boron enolates". J. Am. Chem. Soc. 103: 3099–3111. doi:10.1021/ja00401a031.
- ^ Evans D. A., Rieger D. L., Bilodeau M. T., Urpi F. (1991). "Stereoselective aldol reactions of chlorotitanium enolates. An efficient method for the assemblage of polypropionate-related synthons". J. Am. Chem. Soc. 113: 1047–1049. doi:10.1021/ja00003a051.
- ^ Evans, D. A. et al. Top. Stereochem. 1982, 13, 1–115. (Review)
- ^ Roush W. R. (1991). "Concerning the diastereofacial selectivity of the aldol reactions of .alpha.-methyl chiral aldehydes and lithium and boron propionate enolates". J. Org. Chem. 56: 4151–4157. doi:10.1021/jo00013a015.
- ^ Masamune S., Ellingboe J. W., Choy W. (1982). "Aldol strategy: coordination of the lithium cation with an alkoxy substituent". J. Am. Chem. Soc. 104: 1047–1049. doi:10.1021/ja00384a062.
- ^ a b Evans D. A., Dart M. J., Duffy J. L., Rieger D. L. (1995). "Double Stereodifferentiating Aldol Reactions. The Documentation of "Partially Matched" Aldol Bond Constructions in the Assemblage of Polypropionate Systems". J. Am. Chem. Soc. 117: 9073–9074. doi:10.1021/ja00140a027.
- ^ Masamune S., Choy W., Petersen J. S., Sita L. R. (1985). "Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis". Angew. Chem. Int. Ed. Engl. 24: 1–30. doi:10.1002/anie.198500013.
- ^ Evans, D. A. Aldrichimica Acta 1982, 15, 23. (Review)
- ^ Gage, J. R.; Evans, D. A. Organic Syntheses, Coll. Vol. 8, p.339 (1993); Vol. 68, p.83 (1990). (Article)
- ^ Evans D. A., Bartroli J., Shih T. L. (1981). "Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates". J. Am. Chem. Soc. 103: 2127–2129. doi:10.1021/ja00398a058.
- ^ a b Evans D. A., Bender S. L., Morris J. (1988). "The total synthesis of the polyether antibiotic X-206". J. Am. Chem. Soc. 110: 2506–2526. doi:10.1021/ja00216a026.
- ^ Evans D.A., Clark J.S., Metternich R., Sheppard G.S. (1990). "Diastereoselective aldol reactions using .beta.-keto imide derived enolates. A versatile approach to the assemblage of polypropionate systems". J. Am. Chem. Soc. 112: 866–868. doi:10.1021/ja00158a056.
- ^ Evans D.A., Ng, H.P., Clark J.S., Rieger D.L. (1992). "Diastereoselective anti aldol reactions of chiral ethyl ketones. Enantioselective processes for the synthesis of polypropionate natural products". Tetrahedron. 48: 2127–2142. doi:10.1016/S0040-4020(01)88879-7.
- ^ Dalam reaksi ini, nukleofilnya adalah turunan enolat boron dari reaksi dibutilboron triflat dan basanya adalah N,N-Diisopropiletilamina. Tioeter dilepaskan pada langkah kedua menggunakan Nikel Raney / reduksi hidrogen
- ^ Teruaki Mukaiyama, Kazuo Banno, and Koichi Narasaka (1974). "Reactions of silyl enol ethers with carbonyl compounds activated by titanium tetrachloride". J. Am. Chem. Soc. 96 (24): 7503–7509. doi:10.1021/ja00831a019.
- ^ 3-Hydroxy-3-Methyl-1-Phenyl-1-Butanone by Crossed Aldol Reaction Teruaki Mukaiyama and Koichi Narasaka Organic Syntheses, Coll. Vol. 8, p.323 (1993); Vol. 65, p.6 (1987) Link
- ^ Kruger J., Carreira E.M. (1998). "Apparent catalytic generation of chiral metal enolates: Enantioselective dienolate additions to aldehydes mediated by Tol-BINAP center Cu(II) fluoride complexes". J. Am. Chem. Soc. 120: 837–8. doi:10.1021/ja973331t.
- ^ Pagenkopf B.L., Kruger J., Stojanovic A., Carreira E.M. (1998). "Mechanistic insights into Cu-catalyzed asymmetric aldol reactions: Chemical and spectroscopic evidence for a metalloenolate intermediate". Angew. Chem. Intl. Ed. 37: 3124–6. doi:10.1002/(SICI)1521-3773(19981204)37:22<3124::AID-ANIE3124>3.0.CO;2-1.
- ^ Crimmins M. T., King B. W., Tabet A. E. (1997). "Asymmetric Aldol Additions with Titanium Enolates of Acyloxazolidinethiones: Dependence of Selectivity on Amine Base and Lewis Acid Stoichiometry". Journal of the American Chemical Society. 119 (33): 7883–7884. doi:10.1021/ja9716721.
- ^ Crimmins M. T., Chaudhary K. (2000). "Titanium enolates of thiazolidinethione chiral auxiliaries: Versatile tools for asymmetric aldol additions". Organic Letters. 2 (6): 775–777. doi:10.1021/ol9913901.
- ^ Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
- ^ Asymmetric synthesis of bicyclic intermediates of natural product chemistry Zoltan G. Hajos, David R. Parrish J. Org. Chem.; 1974; 39(12); 1615-1621. doi:10.1021/jo00925a003
- ^ New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures Angewandte Chemie International Edition in English Volume 10, Issue 7, Date: July 1971, Pages: 496-497 Ulrich Eder, Gerhard Sauer, Rudolf Wiechert doi:10.1002/anie.197104961
- ^ The ying and yang of asymmetric aminocatalysis Benjamin List Chem. Commun., 2006, 819–824, doi:10.1039/b514296m
- ^ The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes Alan B. Northrup and David W. C. MacMillan J. Am. Chem. Soc.; 2002; 124(24) pp 6798–6799; (Communication) doi:10.1021/ja0262378
- ^ Northrup A. B., Mangion I. K., Hettche F., MacMillan D. W. C. (2004). "Enantioselective Organocatalytic Direct Aldol Reactions of -Oxyaldehydes: Step One in a Two-Step Synthesis of Carbohydrates". Angewandte Chemie International Edition in English. 43 (16): 2152–2154. doi:10.1002/anie.200453716.
- ^ Diastereoselective Magnesium Halide-Catalyzed anti-Aldol Reactions of Chiral N-Acyloxazolidinones Evans, D. A.; Tedrow, J. S.; Shaw, J. T.; Downey, C. W. J. Am. Chem. Soc.; (Communication); 2002; 124(3); 392–393. doi:10.1021/ja0119548 10.1021/ja0119548; OL 2002, 4, 1127
- ^ Catalytic Enantioselective Thioester Aldol Reactions That Are Compatible with Protic Functional Groups Magdziak, D.; Lalic, G.; Lee, H. M.; Fortner, K. C.; Aloise, A. D.; Shair, M. D. J. Am. Chem. Soc.; (Communication); 2005; 127(20); 7284–7285. doi:10.1021/ja051759j